Поликистоз почек на УЗИ (лекция на Диагностере)
Почечный поликистоз (поликистозная дегенерация, поликистозная болезнь) — это наследуемая врожденная аномалия, при которой обе почки усеяны множеством кист. Между кистами имеются скудные участки неизмененной паренхимы. Иногда почечный поликистоз сочетается с поликистозными изменениями в печени, селезенке, реже — легких, костях, поджелудочной железе, яичниках и придатке яичка.
Важно!!! Случаи одностороннего поражения являются или мультикистозной дисплазией, или очень рано диагностированным истинным поликистозом, неравномерно развитым в обеих почках.
Нажимайте на картинку, чтобы увеличить.
| Фото. Поликистоз относят к цилиопатиям — это группа заболеваний, когда вследствие аномальной структуры ресничек усилена пролиферация кистозных клеток и секреция жидкости в просвет. | ||
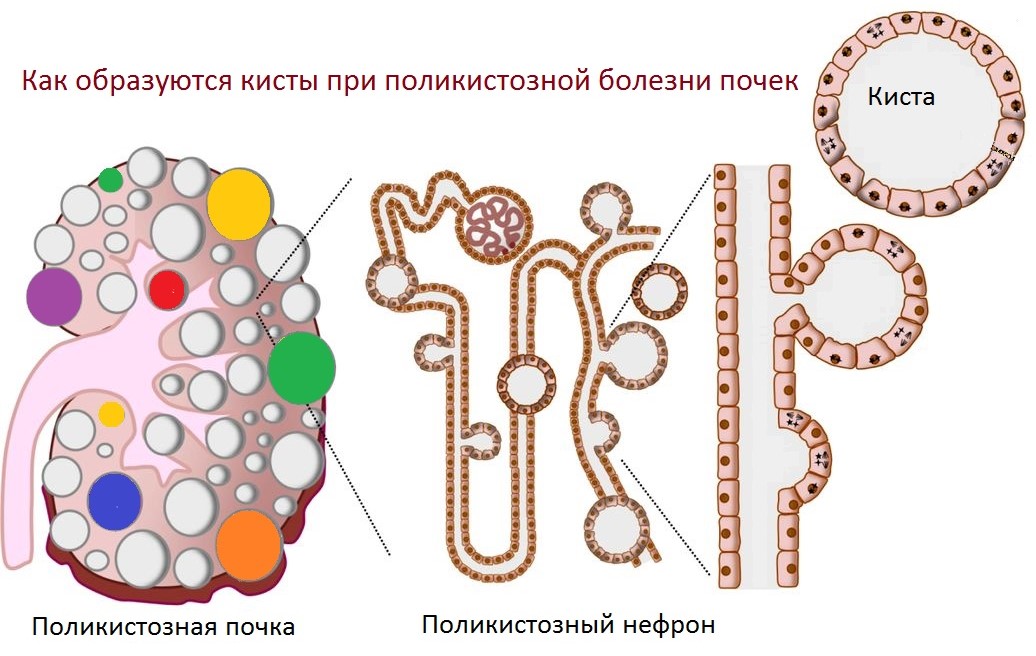 |
||
| Фото. А — При аутосомно-рецессивном поликистозе (АРПКП) кистозно расширенные собирательные трубочки расположены радиально от мозгового вещества к коре. Б — При аутосомно-доминантном поликистозе (АДПКП) кисты образуются во всех сегментах нефрона и быстро теряют связь с ним. | ||
 |
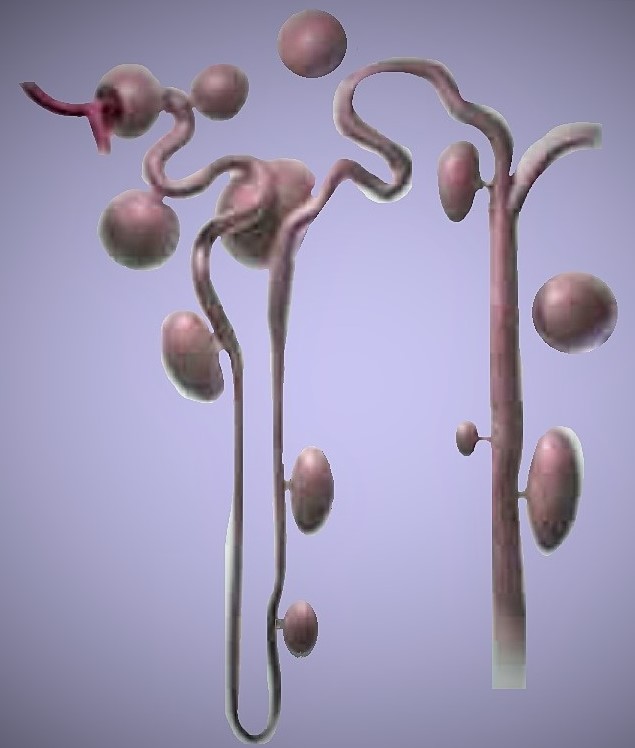 |
|
Аутосомно-рецессивный поликистоз почек на УЗИ
Злокачественный поликистоз детского возраста передается аутосомно-рецессивно. Причиной АРПКП считают мутации гена PKHD1 на хромосоме 6 (6p12). Ген продуцирует белок под названием фиброцистин или полиумин, функция которого не ясна.
| Фото. Как наследуется аутосомно-рецессивный поликистоз почек: оба родителя здоровы, но являются носителями дефектного гена; вероятность рождения больного ребенка в таком браке составляет 25%. | ||
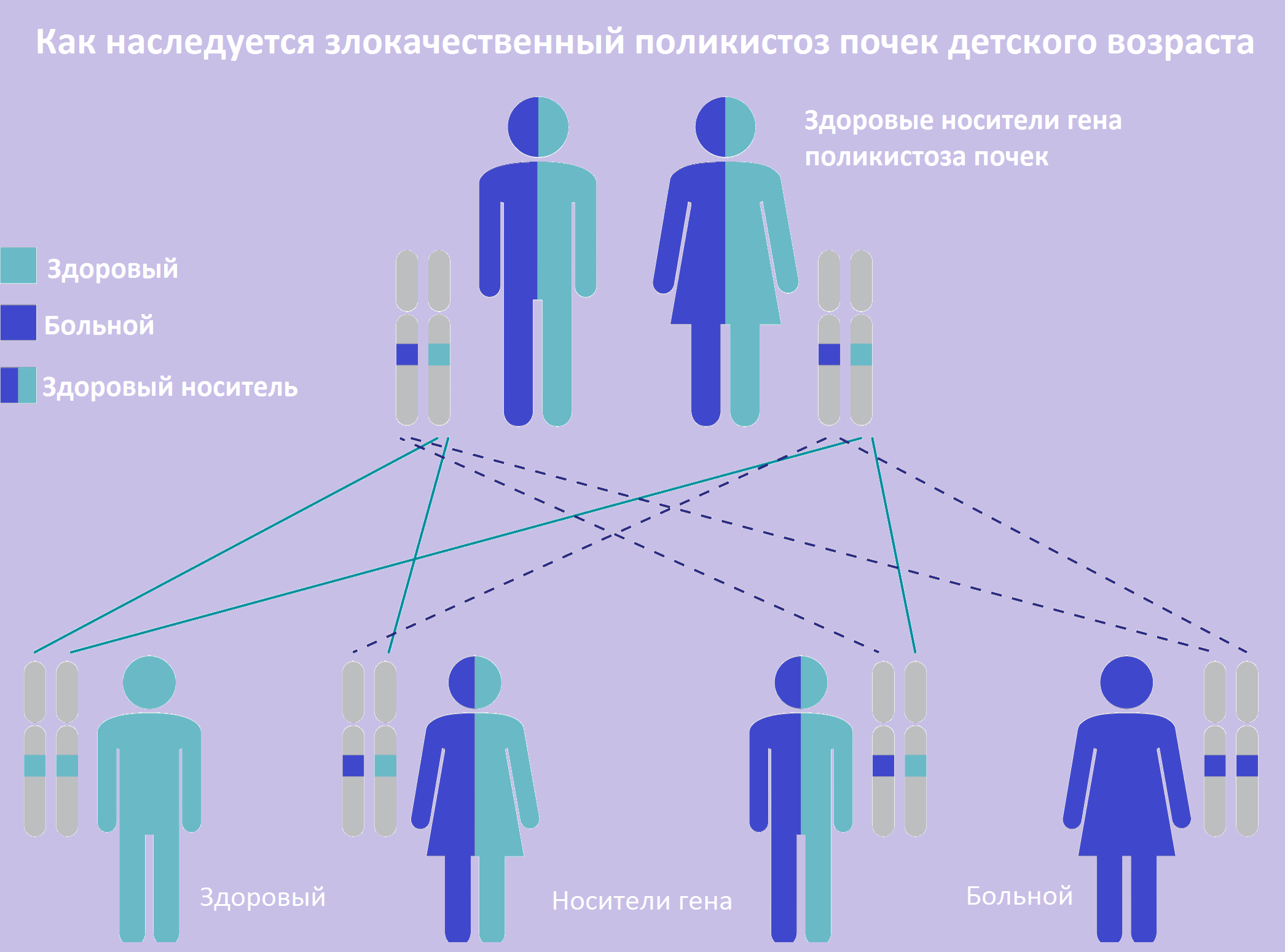 |
||
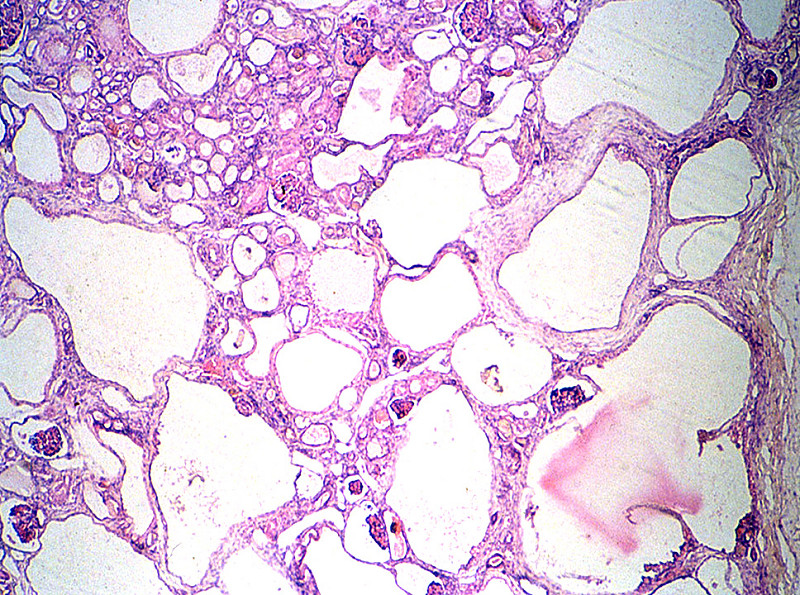
При АРПКП у младенцев обе почки увеличены. По всей паренхиме от мозгового вещества к коре радиально расположены цилиндрические пространства — кисты небольших размеров. Кисты представляют собой расширение собирательных трубочек и сохраняют связь с нефроном. Граница между корковым и мозговым слоем сглажена. При АРПКП всегда имеется разной степени врожденный фиброз печени.
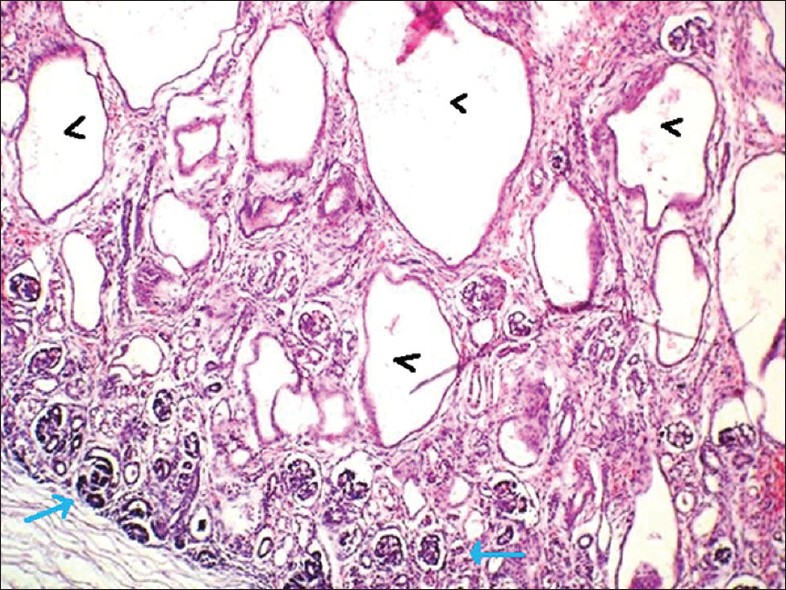
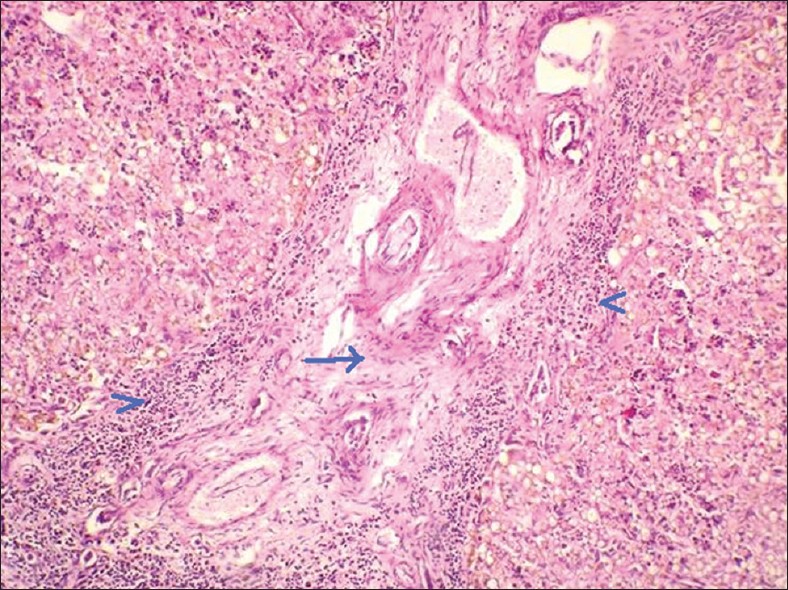
| Фото. А — Почки плода увеличены, на срезе многочисленные кисты в коре и мозговом веществе от 5 до 7 мм (стрелки), нарушена кортикомедулярная дифференцировка. Б — В почках субкапсулярно расположена нефрогенная зона с клубочками (стрелка); ниже в коре и мозговом веществе определяются многочисленные кисты разных размеров, выстланные кубическим эпителием (галочки). В — В печени пролиферация желчных протоков, очаговый фиброз (стрелка), внутрипеченочный холестаз, перипортальная лимфоцитарная инфильтрация (галочки). Диагноз: аутосомно-рецессивная поликистозная болезнь почек. | ||
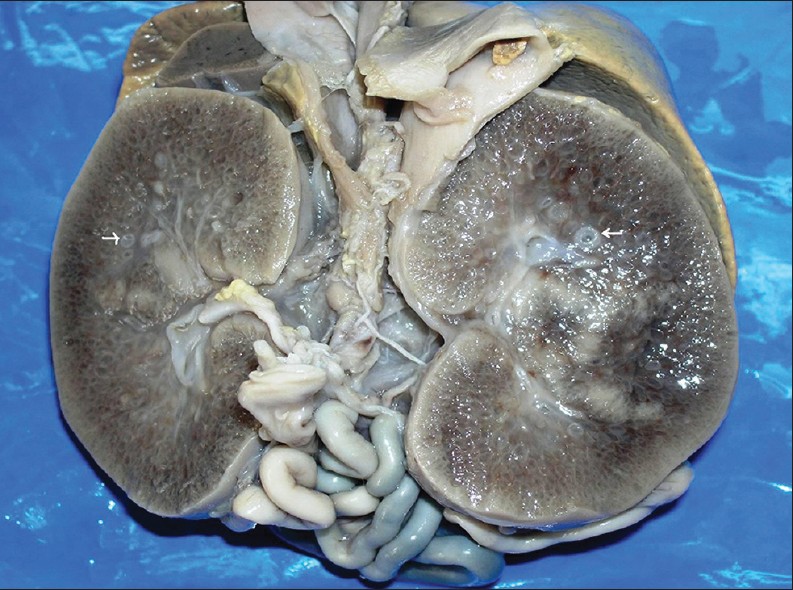 |
 |
 |
АРПКП диагностируют у 1 из 10000-50000 новорожденных. АРПКП делят на перинатальную, неонатальную, инфантильную и ювенильную формы:
Важно!!! Чем раньше проявляется поликистоз, тем злокачественней он протекает и тем хуже прогноз.
- При перинатальной форме АРПКП младенцы рождаются с большими поликистозными почками и умирают в первые дни жизни от уремии или дыхательной недостаточности вследствие гипоплазии легких.
- При неонатальной форме АРПКП более 90% нефронов поражены кистами, быстро прогрессирует почечная недостаточность; фиброз печени слабо выражен.
- При инфантильной форме АРПКП только часть нефронов кистозно изменены, почечная недостаточность развивается ближе к подростковому возрасту; портальная гипертензия и гиперспленизм вследствие фиброза печени редко бывают ведущими симптомами.
- При ювенильной форме АРПКП кистозно расширены менее 10% нефронов, почечная недостаточность обычно отсутствует, НО к 20-ти годам появляются признаки портальной гипертензии вследствие фиброза печени.
АРПКП можно заподозрить при УЗИ плода: гиперэхогенные почки сильно больших размеров, мочевой пузырь часто не определяется; характерно маловодие; в стесненных условиях невозможно нормальное развитие легких плода, поэтому формируется гипоплазия легких.
Важно!!! Плоское лицо, косолапость и другие деформации конечностей, гипоплазия легких у младенцев с дисгенезией почек называют синдромом Поттера. Считается, что причиной этих нарушений является маловодие вследствие отсутствия нормальной продукции мочи у плода с патологией почек.
Важно!!! Большие почки у плода бывают при синдромах генерализованного избыточного роста — синдром Перлмана и синдром Бэквита-Видемана.
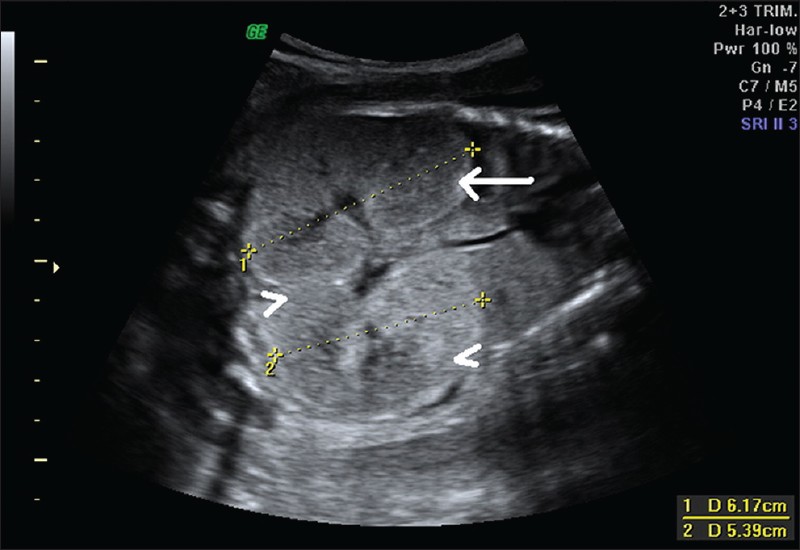
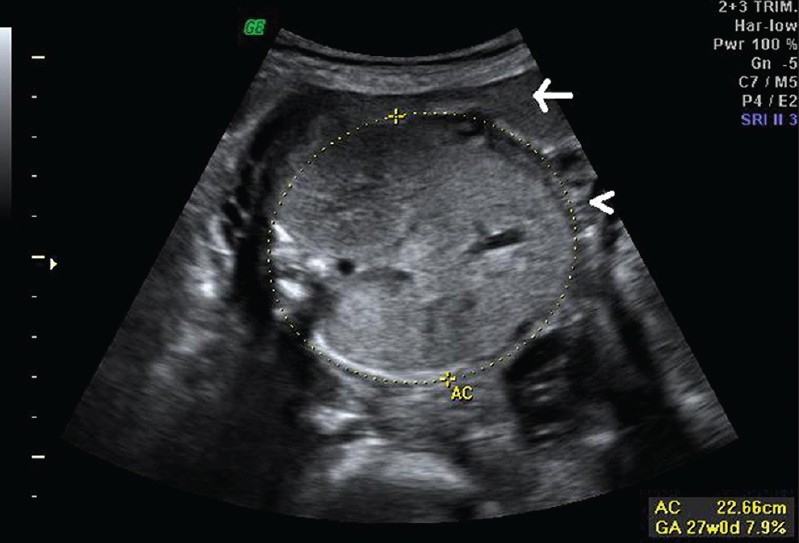
| Фото. На УЗИ женщина 26-ти лет во втором триместре беременности: А — Почки плода значительно увеличены, гиперэхогенные (стрелка), кортикомедуллярная дифференцировка сглажена (галочки). Б — Объем брюшной полости плода увеличен, желудок и мочевой пузырь не определяются, переднее расположение плаценты (стрелка), выраженное маловодие (галочка). В — Плацента утолщена до 6,1 см. Диагноз: выраженное маловодие, аутосомно-рецессивная поликистозная болезнь плода. Беременность была прервана, извлечен плод мужского пола весом 900 г. | ||
 |
 |
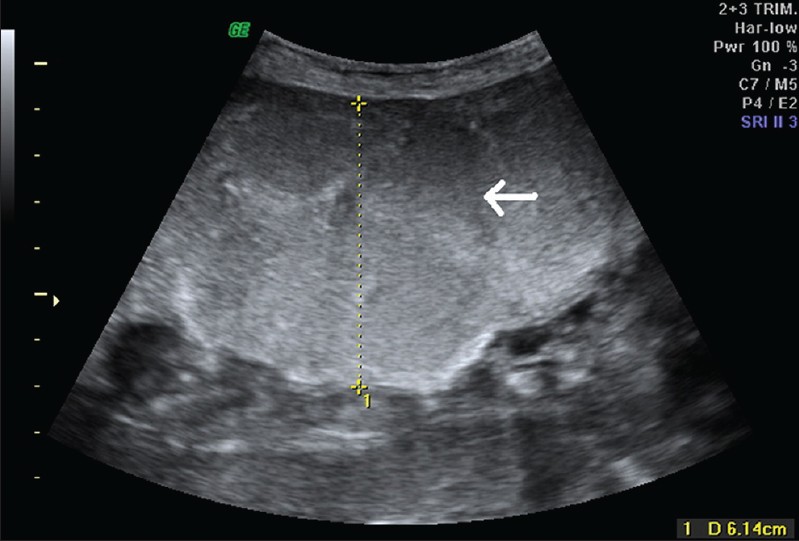 |
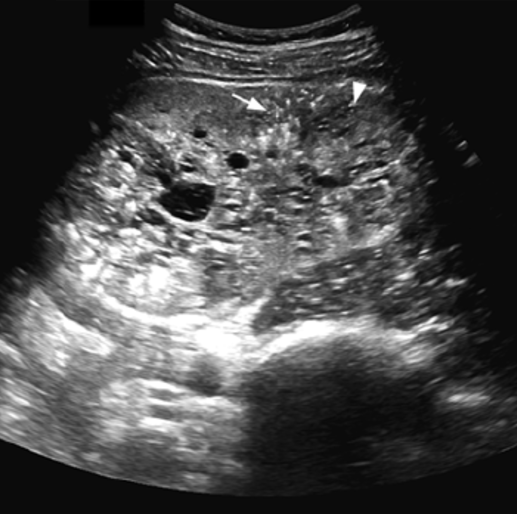
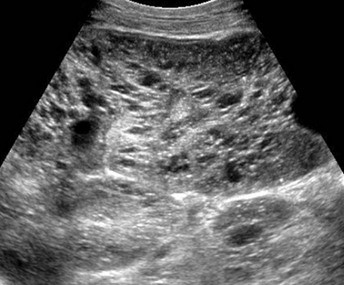
При аутосомно-рецессивном поликистозе на УЗИ обе почки симметрично увеличены, многочисленные крошечные кисты обычно не определяются ультразвуком, но создают картину неоднородной гиперэхогенной почки без признаков кортикомедулярной дифференцировки. Тонкий гипоэхогенный ободок по периферии представляет собой сжатую кору. Кроме того, следует обращать внимание на признаки портальной гипертензии, холангиопатии, дисгенезии желчных путей.
Важно!!! При мягкой ювенильной форме АРПКП и на ранних стадиях АДПКП на УЗИ кисты в почках могут выглядеть одинаково. В таких случаях наличие фиброза печени или болезни Кароли указывает на АРПКП, а при АДПКП следует искать поликистоз почек у одного из родителей.
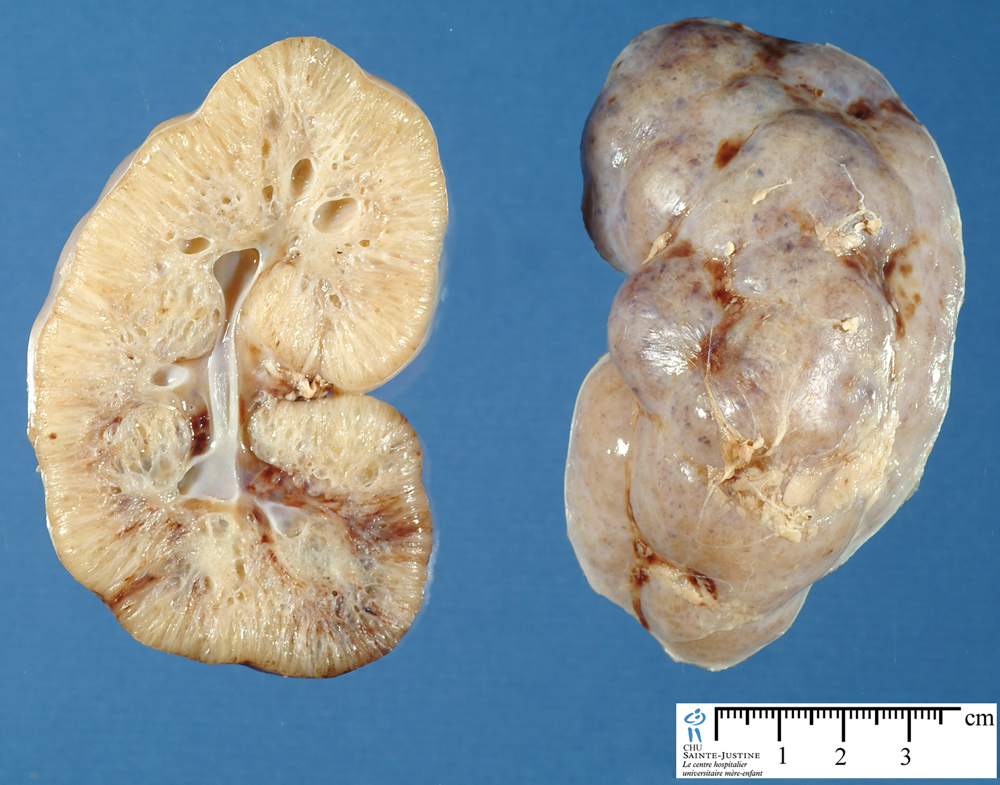
| Фото. А — Почки младенца с АРПКП: большие белые почки с выраженной дольчатостью, множество мелких кист придают им губчатый вид, граница между корковым и мозговым слоем сглажена. Б, В — Ребенок в возрасте 1 года с АРПКП и врожденным фиброзом печени: на УЗИ почки увеличены, неоднородные за счет множества точечных гиперэхогенных очагов с мелкими кистами, кортикомедуллярная дифференцировка сглажена. | ||
 |
 |
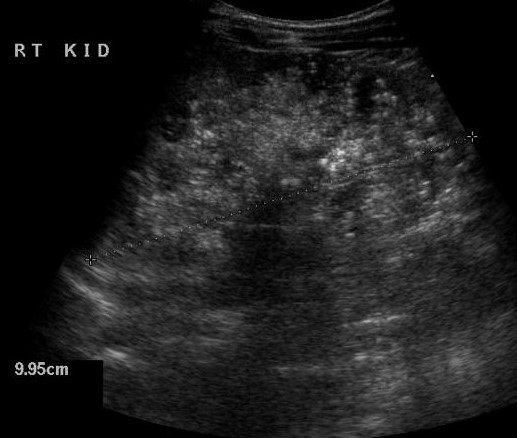 |
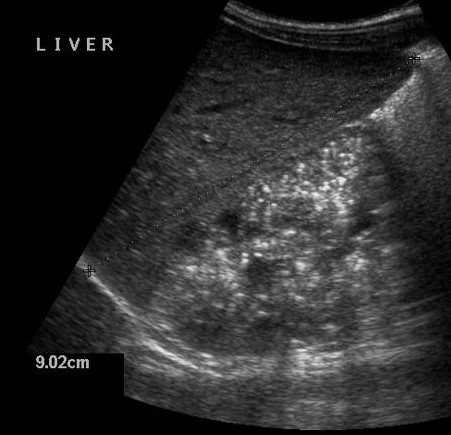
| Фото. А — Печень и почки младенца с АРПКП: хорошо заметно, что почки сильно большие. Б, В — АРПКП у младенцев с почечной недостаточностью: на УЗИ почка увеличена, гиперэхогенная за счет ярких включений с хвостом кометы (стрелка) и расширенными канальцами (треугольник), перенхима губчатой эхоструктуры, кортикомедуллярная дифференциация отсутствует. | ||
 |
 |
 |
Аутосомно-доминантный поликистоз почек на УЗИ
Взрослый тип поликистоза передается аутосомно-доминантно. В 90% случаев удается проследить семейный характер заболевания, НО 10% случаев возникает спорадически. Причиной АДПКП считают мутацией в генах PDK1, PDK2 и PDK3:
- Мутации в гене PDK1 составляют 85% случаев АДПКП. Ген PDK1 на коротком плече 16 хромосомы (16p13.3) кодирует белок полицистин-1. Кисты в почках определяются к 10 годам у 64% больных и к 20 годам — у 90%. Почечная недостаточность, как правило, развивается к 50-ти годам.
- Мутации в гене PDK2 составляют около 15% случаев АДПКП. Ген PKD2 на длинном плече 4 хромосомы (4q21-23) кодирует белок полицистин-2. У этих пациентов болезнь прогрессирует медленнее, и почечная недостаточность развивается к 70-ти годам.
- Когда у больного АДПКП не находят мутации в генах PDK1 и PDK2, говорят о мутации в гене PDK3, который расположен в неизвестной хромосоме.
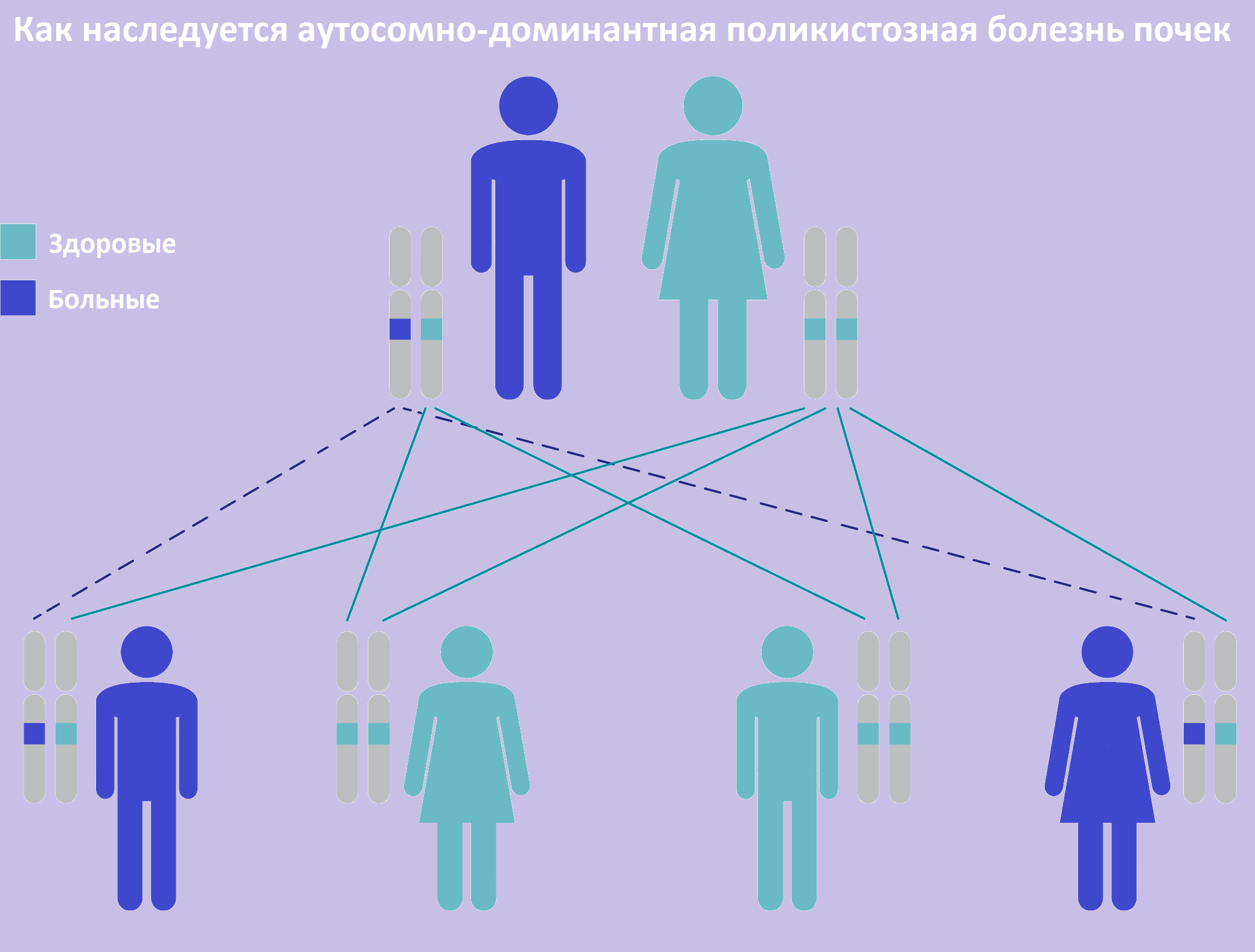
| Фото. Как наследуется аутосомно-доминантная поликистозная болезнь почек: так как поликистоз передается как аутосомно-доминантный признак, вероятность рождения больного ребенка в семье, где один из родителей болен, составляет 50%. | ||
 |
||
Критерии диагноза при наличие АДПКП у одного из родителей:
- две односторонние или двусторонние кисты до 30 лет;
- две кисты в каждой почке у лиц в возрасте от 30 до 59 лет;
- четыре кисты в каждой почке в возрасте 60 лет и старше.
Аутосомно-доминантная поликистозная болезнь почек (АДПКП) встречается с частотой 1 случай на 400-1000 человек. Среди пациентов с хронической почечной недостаточностью от 7% до 15% — это больные с АДПКП.
АДПКП обычно НЕ распознают внутриутробно, потому что почки выглядят нормальными, сохранена кортикомедуллярная дифференцировка, присутствует мочевой пузырь и объем амниотической жидкости адекватный. В очень редких случаях при АДПКП новорожденные уже имеют крупные кисты, или их почки выглядят большими и гиперэхогенными, как при АРПКП. В таких ситуациях нужно искать родителя с АДПКП для подтверждения диагноза. До сих пор все случаи АДПКП у плода были связаны с мутацией в гене PKD1.
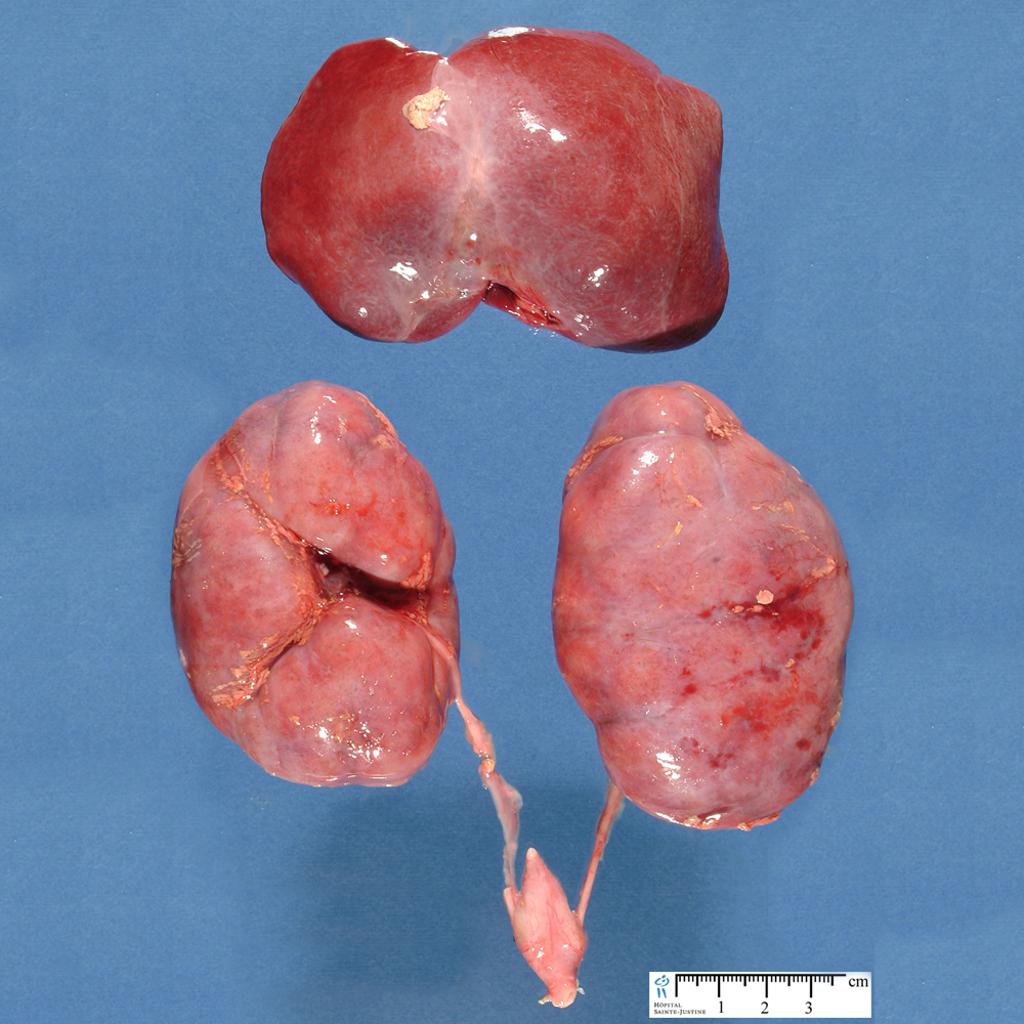
| Фото. Поликистоз почек у новорожденного мальчика. | ||
 |
 |
 |
Первые клинические признаки АДПКП появляются после 15 лет: тупые боли в пояснице, «немотивированная» периодическая гематурия, полиурия с гипостенурией и никтуриея. К 35-40 годам обычно развивается умеренная и медленно прогрессирующая почечная недостаточность. Часто наблюдается гипертония (70%), обычно доброкачественная. Иногда удается прощупать сильно увеличенные бугристые почки. В возрасте 40 лет состояние обычно начинает быстро ухудшаться, нарастает водно-солевая декомпенсация с явлениями обезвоживания, гиперазотемии или гипонатриемии. Позднее наступают гиперфосфатемия с гипокальциемией, ацидоз с уменьшением щелочного резерва. Тем не менее при адекватном солево-водном режиме многие больные могут поддерживать сбалансированное состояние годами. Острые лихорадочные заболевания резко нарушают водно-электролитное равновесие и выделительные возможности. Смертельный исход наступает обычно через 10-12 лет после появления первых клинических признаков, однако многие больные живут и больше 70 лет, когда смерть наступает от почечной недостаточности или от другого заболевания.
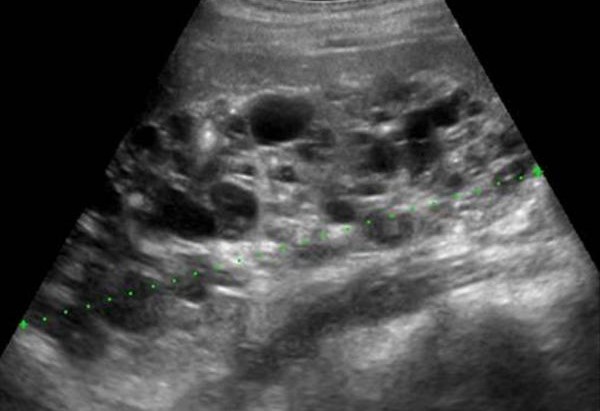
| Фото. Мальчик 11-ти лет с отягощенной наследственностью — поликистоз почек у матери. Из анамнеза: В возрасте 3-х лет на УЗИ впервые отмечают повышенную эхогенность паренхимы почек. В 6 лет на УЗИ: паренхима повышенной эхогенности с обеих сторон, неоднородная за счет гиперэхогенных включений и множественных диффузно расположенных мелких кист (2-4 мм) в корковом слое и субкапсулярно. На фоне ОРВИ несколько эпизодов уреженного мочеиспускания (до 2-х раз в сутки маленькими порциями) с отеками на голенях и одутловатостью лица. Диагноз: поликистоз почек по аутосомно-доминантному типу. А, Б — Обратите внимание, кисты в паренхиме почки очень мелкие (2-4 мм) и определяются с трудом. Очень важно оценить почку в сравнении с печенью, тогда бросается в глаза гиперэхогенная по отношению к печени паренхима почки. В норме паренхима почки почти всегда темнее печени. | ||
 |
 |
|
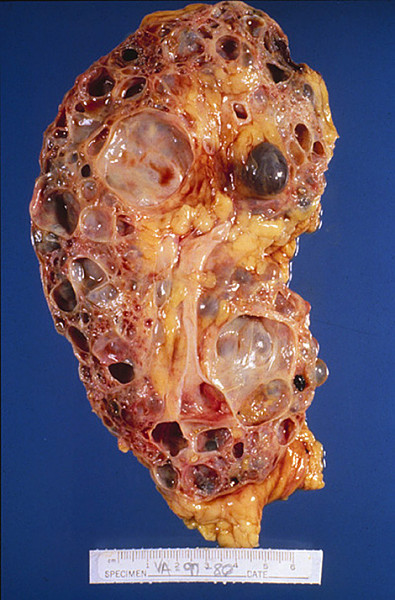
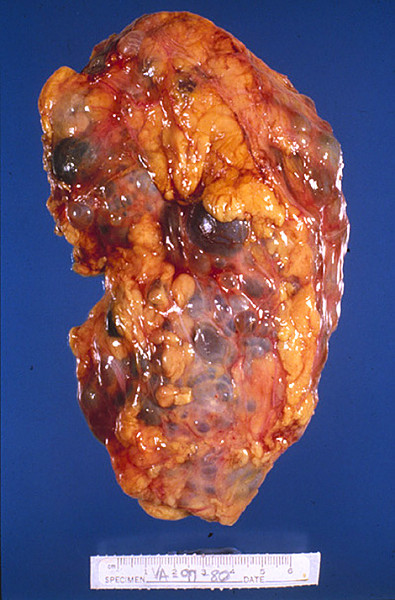
У взрослых с АДПКП почки представляют собой огромную деформированную массу с множественными круглыми кистами различного размера — от просяного зерна до апельсина. При АДПКП кисты образуются во всех сегментах нефрона, распределяются по всей паренхиме почки и быстро теряют связь с канальцем. Лоханка и чашечки значительно удлинены и деформированы; кисты выстланы кубовидным или плоским эпителием, а стенки — фибро-коллагенные. Между кистами находятся скудные количества паренхимы, с дегенерацией в результате сдавления, но без диспластических изменений. Как правило, в имеющейся паренхиме обнаруживаются вторичные изменения типа интерстициального нефрита. В некоторых случаях кисты бывают настолько многочисленны и велики, что паренхима почти полностью отсутствует. Кистозная жидкость — водянистая с низким удельным весом (1004-1006), желтоватая или коричневатая. Состав кистозной жидкости дает основания считать, что в кистах содержится моча. Иногда, после вторичных изменений, она становится желто-слизистой или кровянистой.
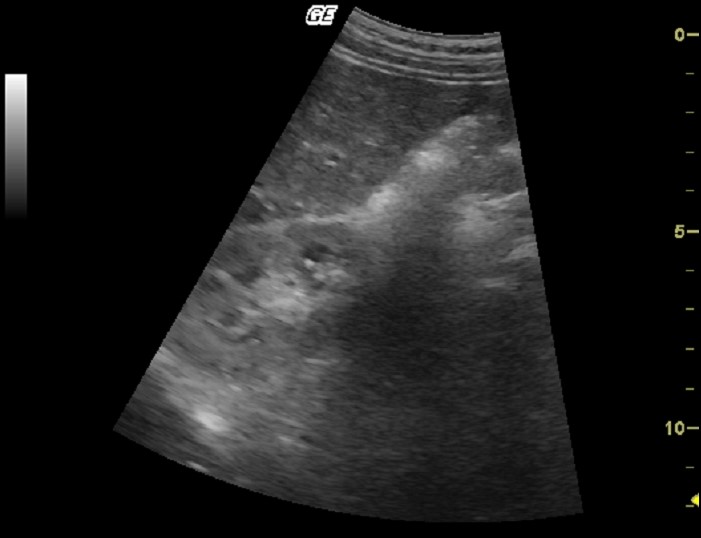
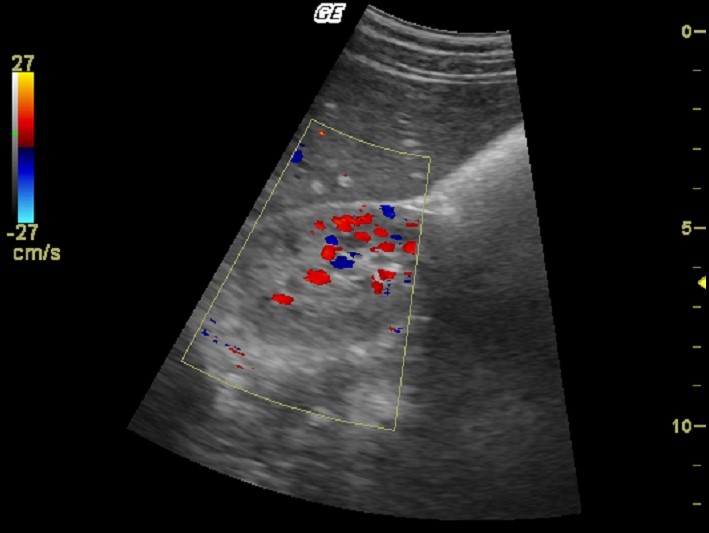
| Фото. Пациент с терминальной стадией почечной недостаточности вследствие АДПКП. Поликистозная почка на УЗИ сильно больших размеров, множественные округлые кисты определяются во всех отделах паренхимы. Произведена трансплантация почки. | ||
 |
 |
|
Поликистозные почки особенно подвержены другим заболеваниям и осложнениям: туберкулезу, пиелонефриту, гидронефрозу, пионефрозу, мочекаменной болезни. Осложнения резко ухудшают это спокойное медленное течение заболевания. Камни в почках вызывают почечные колики, могут быть причиной немотивированной гематурии. Для поликистоза характерна гематурия и без наличия камней, но она наступает редко, обычно через месячные промежутки. Быстрое нарастание кист вследствие кровоизлияния или нагноения может причинить постоянные боли в пояснице, а также и задержку мочи. Если другая почка не функционирует, то возникает острая анурия. Нагноение кисты или пионефроз проявляется болями в пояснице и картиной уросепсиса. Пионефроз может осложниться паранефритом. Заболевание имеет тяжелое течение и плохой прогноз. Острая олигурия и анурия может наступить в результате недостаточной коррекции потери воды при рвотах, поносах и пр. При таких явлениях следует своевременно принимать меры. При продолжительном течении могут развиться вторичные изменения костно-суставного аппарата и нервной системы. В некоторых случаях развивается подагрозный синдром, обусловленный понижением клиренса мочевой кислоты. Гипокальциемия в результате хронической гиперкальциурии иногда вызывает костную деминерализацию со спонтанными переломами, так же как и нервно-мышечные судороги. Последние наступают обычно в связи с гиперкоррекцией ацидоза щелочами. Реже развивается уремический полиневрит.
Печеночные кисты — это наиболее распространенное внепочечное проявление АДПКП. Они редки у детей, и их частота увеличивается с возрастом. В возрасте 50-ти лет кисты печени имеются практически у всех пациентов с АДПКП. Кисты печени обычно бессимптомны, но в редких случаях большие кисты приводят к портальной гипертензии и кровотечению из варикозных вен пищевода. Если появляется вторичная портальная гипертензия, отличить АРПКП от АДПКП трудно. При АРПКП портальная гипертензия встречается гораздо чаще и всегда вторична по отношению к врожденному фиброзу печени.
Внутричерепные аневризмы — преимущественно аневризмы Виллизиева круга — обнаруживают у 10-30% пациентов с АДПКП, и приблизительно 9% из них умирают из-за субарахноидальных кровоизлияний.
Берегите себя, Ваш Диагностер!